Friedreich Ataxia; Shkaqet, Simptomat, Prognoza, Trajtimi

Friedreich Ataxia; Shkaqet, Simptomat, Prognoza, Trajtimi
Friedreich Ataxia (FA) është një sëmundje e rrallë, e trashëguar dhe progresive e nervave dhe e muskujve. E quajtur sipas emrit të shkencëtarit gjerman që e përshkroi atë së pari, FA shkakton humbjen e kontrollit të lëvizjeve të trupit (ataxia). Zakonisht fillon në fëmijëri ose adoleshencë dhe përkeqësohet me moshën. Shenja e parë e FA është zakonisht vështirësia në ecje për shkak të humbjes së koordinimit vullnetar të muskujve të këmbëve. Një person me FA zakonisht ka nevojë për një karrocë invalidësh brenda 10 viteve të fillimit të simptomave, mund të jetë plotësisht i paaftë nga mosha e mesme dhe mund të ketë një jetëgjatësi më të shkurtër.
Shkaqet e Ataxisë së Friedreich-it
Friedreich Ataxia (FA), një gjendje gjenetike që ndikon në sistemin nervor dhe muskujt, shkaktohet nga një mutacion në një gjen të quajtur furaxin (FXN). Gjeni FXN jep udhëzime për proteinën frataxin, e cila është thelbësore për funksionimin e duhur të mitokondriteve, fabrikave të prodhimit të energjisë brenda qelizës.
- Shkaku gjenetik i FA
Dëmi gjenetik që shkakton Friedreich Ataxia është një përsëritje e rritjes së trinukleotideve GAA. Secili gjen përbëhet nga një sekuencë koduese e përbërë nga katër njësi kimike të quajtura nukleotide – adeninë (A), citozinë (C), guaninë (G) dhe timinë (T).
Në gjenin FXN, ekziston një rajon ku tre nukleotide, GAA, përsëriten disa herë (nga 5 deri në 33 herë). Numri i përsëritjeve në njerëzit me FA është i rritur (duke filluar nga 66 përsëritje në më shumë se 1.000 përsëritje) në të dy kopjet e gjenit FXN.
Gjatësia e përsëritjes së trinukleotideve GAA duket të jetë e lidhur me moshën e fillimit, shkallën e ashpërsisë dhe ritmin e përparimit të simptomës. Njerëzit me segmente të GAA-s të përsëritura më pak se 300 herë tentojnë të kenë një simptomë më të vonshme (pas moshës 25 vjeç) sesa ato me trinukleotide më të mëdha të GAA-s.
Rreth 2% e njerëzve me FA kanë një përsëritje të trinukleotideve GAA të zgjeruar në një kopje dhe një lloj tjetër mutacioni në kopjen tjetër të gjenit FXN. Në shumicën e këtyre rasteve, mutacioni tjetër ndryshon një nukleotid kodues të vetëm (të quajtur një pikë mutacion), brenda gjenit FXN.
Edhe pse nuk është e qartë se si këto mutacione shkaktojnë Friedreich Ataxia, shkencëtarët mendojnë se proteina e frakaksinës kontrollon prodhimin e energjisë në mitokondri. Frataxin mban hekurin në një formë të lidhur dhe e siguron atë për sintezën e enzimeve mitokondriale të përdorura për të prodhuar energji. Kur ka mungesë të frataxin, i gjithë hekuri është në formë të lirë, dhe nuk mund të përdoret për sintezën e enzimës mitokondriale, duke rezultuar në një përçarje në prodhimin e energjisë. Hekuri i lirë pastaj grumbullohet, duke rezultuar në një grumbullim të molekulave të dëmshme reaktive (të quajtura radikale të lira), duke dëmtuar dhe shkatërruar qelizat.
Meqenëse mitokondria është një burim thelbësor i energjisë, qelizat në tru, palcë kurrizore dhe muskujt, të cilat kërkojnë shumë energji, janë veçanërisht të ndjeshme ndaj problemeve mitohondriake dhe mund të dëmtohen lehtësisht nga një furnizim i pamjaftueshëm me energji. Kjo çon në shenjat dhe simptomat e Friedreich Ataxia.
- Trashëgimia
FA trashëgohet në një mënyrë autosomale recesive, që do të thotë se një individ zhvillon FA vetëm kur ai ose ajo mbart defekte gjenetike në të dy kopjet e gjenit FXN: një kopje e trashëguar nga secili prind.
Nëse një individ trashëgon një kopje normale dhe një kopje me defekt të gjenit FXN, ai ose ajo nuk tregon simptoma dhe thuhet se është një transportues. Nëse të dy prindërit janë transportues, rreziku i një fëmije që zhvillon FA është 25%.
Simptomat e Friedreich Ataxia
Simptomat e Friedreich Ataxia zakonisht fillojnë në mes të moshave 5 dhe 15 vjeç. Individët me një numër të vogël të përsëritjeve (më pak se 300) kanë tendencë të kenë një sëmundje të vonë fillestare (pas moshës 25 vjeçare). Në raste të rralla, shfaqja e sëmundjes mund të jetë deri në moshën 75 vjeçare.
Megjithëse shkalla e përparimit ndryshon, një individ me FA ka nevojë për karrocë brenda 10 deri në 20 vjet pas paraqitjes së simptomave. Në fazat e mëvonshme të sëmundjes, individët mund të bëhen plotësisht të dobësuar.
FA mund të shkurtojë jetëgjatësinë me sëmundjen e zemrës duke qenë shkaku më i zakonshëm i vdekjes. Megjithatë, disa njerëz me një sëmundje më pak të rëndë mund të jetojnë deri në të gjashtëdhjetat, shtatëdhjetat e tyre apo edhe më shumë.
Edhe pse aftësitë mendore të njerëzve me FA mbeten krejtësisht të paprekura, humbja progresive e koordinimit dhe fuqia e muskujve çon në paaftësi motorike dhe nevojën për të përdorur një karrocë. Shumica e pacientëve të rinj me FA (adoleshentë ose ata në të 20-at e tyre) kërkojnë ndihmë për lëvizshmërinë, si një shkop, patericë ose karrocë.
FA prek shumë organe dhe kështu prodhon një sërë simptomash
- Simptomat neuromuskulare
Këto përfshijnë humbjen e koordinimit ose ataksi në krahë dhe këmbë që përfshijnë lëvizje të mprehta dhe lëkundëse. Tërheqja e ataksisë ose vështirësia në këmbë është zakonisht simptoma e parë që shfaqet. Kjo përkeqësohet gradualisht dhe përhapet në krahë dhe trung. Simptoma të tjera mund të përfshijnë vështirësi balancimi, paralizë e muskujve të këmbëve, vështirësi në lëvizjen e krahëve dhe humbje e ndjesisë (sidomos vibrimet dhe ndjenjën e pozicionit) në gjymtyrë. Karakteristika të tjera përfshijnë humbjen e reflekseve në gjunjë dhe këmbë.
- Deformimet skeletore
Deformimet skeletore shkaktohen nga probleme neuromuskulare. Ato përfshijnë scoliosis agresive (lakimi i shtyllës së shpinës në njërën anë), harkimi i madh i këmbëve, deformimet e këmbëve dhe inversionet e këmbëve (këmbët që kthehen në brendësi). Scoliosis shpesh kërkon ndërhyrje kirurgjikale.
- Problemet neurologjike
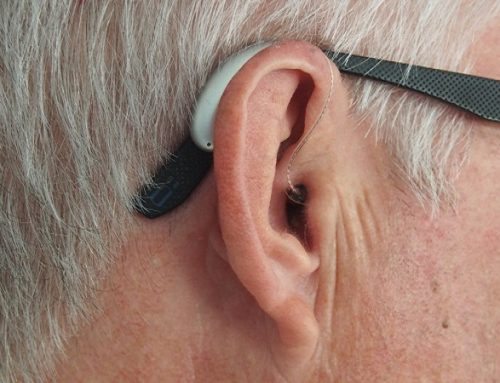
Problemet neurologjike të lidhura me FA përfshijnë vështirësi në të folur ose shqiptim të keq të fjalëve e quajtur “dysarthria”. Ajo përfshin modele të të folurit të ngadalshëm dhe të ngurtaë, që merr progresivisht të bëhet më keq. Probleme të tjera përfshijnë lëvizje të shpejta, të pavullnetshme dhe të mprehta të syrit (nystagmus), vizion të reduktuar dhe humbje të dëgjimit.
- Problemet e zemrës
Në 75% të njerëzve me FA, zhvillohen anomalitë në zemër. Forma të ndryshme të sëmundjeve të zemrës që shoqërojnë FA, përfshijnë cardiomyopathy hypertrophic (zgjerimin dhe dobësimin e muskujve të zemrës), fibrozë miokardit (formimin e material fibroz në muskujt e zemrës), dhe dështim të zemrës. Gjithashtu janë të zakonshme anomalitë e ritmit të zemrës si takikardia (shkalla e shpejtë e zemrës) dhe bllokimi i zemrës (zhvillimi i dëmtuar i impulseve kardiake në zemër).
Këto probleme të zemrës çojnë në simptoma të tilla si lodhje ekstreme, dhimbje gjoksi, frymëmarrje e shkurtër (sidomos me tendosje), dhe rrahje zemre të parregullta (palpitations). Simptomat e dështimit të zemrës përfshijnë ënjtjen e këmbëve dhe vështirësi në frymëmarrjes derisa shtrihen përtokë.
- Diabeti
Rreth 20% e njerëzve me FA zhvillojnë intolerancën e karbohidrateve dhe 10% zhvillojnë diabet. Simptomat e diabetit përfshijnë etje ekstreme, urinim të shpeshtë, humbje peshe, lodhje dhe vizion të paqartë.
Diagnoza e Friedreich Ataxia
Diagnoza e Friedreich Ataxia mund të bëhet me kujdes duke shqyrtuar simptomat, historinë mjekësore dhe çdo histori të çrregullimeve neuromuskulare të familjes. Një mjek do të kryejë një ekzaminim të plotë fizik, veçanërisht të zemrës, dhe ekzaminimin neurologjik, sidomos të këmbëve, krahëve dhe syve.
Testet që mund të ndihmojnë në diagnostikimin e FA përfshijnë studimet e përçimit të nervit, elektromiogram, elektrokardiogramë, echocardiogram, imazhet e rezonancës magnetike (MRI) dhe teste të gjakut. Por një diagnozë përfundimtare e Friedreich Ataxia mund të bëhet vetëm përmes testimit gjenetik.
Prognoza e Friedreich Ataxia
Friedreich Ataxia (FA) është një çrregullim i rrallë gjenetik i trashëguar. Ajo shfaqet në formën e dëmtimit progresiv të koordinimit të muskujve (ataxia), humbjes së forcës dhe ndjeshmërisë së muskujve, dëmtimit të të folurit, vizionit dhe dëgjimit. Sëmundja karakterizohet nga një progresion i ngadalshëm dhe, në përgjithësi, ka një prognozë tepër të dobët. Parashikimi individual varet nga shumë faktorë, duke përfshirë ndryshimet specifike gjenetike sipas gjendjes, moshës së shfaqjes së sëmundjes, ashpërsisë së simptomave dhe pranisë së sëmundjeve të tjera.
Faktorët që ndikojnë në prognozën e Friedreich Ataxia, përfshijnë moshën e fillimit të sëmundjes, ashpërsinë e simptomave dhe komorbiditetet.
- Mosha e fillimit
Mosha e fillimit të sëmundjes ndryshon ndjeshëm mes pacientëve. Shumica e pacientëve (75-85%) janë diagnostikuar me FA para moshës 25 vjeç. Fillimi në një moshë shumë të re (para moshës 5 vjeç) është jashtëzakonisht e rrallë. Rreth 25% e pacientëve kanë një prezantim atipik me fillimin e sëmundjes në moshat më të vjetra. Përafërsisht 15% e rasteve janë diagnostikuar në mes të moshave 26 dhe 39 (të quajtur vonesë e Friedneich’s ataxia (LOFA)), dhe 12% e rasteve janë diagnostikuar pas moshës 40 vjeçare. Kjo quhet Friedreich ataxia shumë e vonë (VLOFA).
Mosha e fillimit dhe prognoza lidhet me numrin e përsëritjeve të trinukleotideve. Pacientët me një numër më të vogël të përsëritjeve (më pak se 300) që zhvillojnë sëmundjen më vonë në jetë, kanë më pak simptoma të rënda dhe zakonisht jetojnë më gjatë.
- Jetëgjatësia
Në përgjithësi, mosha e sëmundjes korrespondon me jetëgjatësinë. Njerëzit me sëmundje të hershme fillojnë të shfaqin simptoma më të rënda dhe vdesin më të rinj. Shumica e pacientëve me FA mbijetojnë deri në moshën 40-50 vjeç, edhe pse jetëgjatësia ndryshon në mënyrë të konsiderueshme në varësi të ashpërsisë së simptomave. Njerëzit me LOFA dhe VLOFA priren të kenë simptoma të buta dhe të mbijetojnë më gjatë.
- Sëmundje dhe komplikime të tjera
Problemi më i zakonshëm (dhe shkaku i vdekjes në 59% të pacientëve) është mosfunksionimi i zemrës, aritmia dhe dështimi kongjestion i zemrës. Pacientët me disfunksione kardiake priren të vdesin më herët – mesatarisht, 17 vjet pas fillimit të sëmundjes. Pacientët pa probleme zemre të lidhura me sëmundjen jetojnë më gjatë dhe mbijetojnë, mesatarisht, për 29 vjet pas fillimit të sëmundjes.
Rreth 10 % e pacientëve me FA zhvillojnë diabet. Ka edhe komplikime të tjera të mundshme që lidhen me sëmundjen, duke përfshirë skoliozionin (lakim të shpinës) dhe problemet me tretjen e karbohidrateve. Prania e komplikimeve gjithashtu mund të ndikojë në prognozën e pacientëve.
Një shumicë dërrmuese (95 %) e pacientëve humbasin aftësinë për të ecur (mesatarisht, 15 vjet pas fillimit të sëmundjes) dhe duhet të përdorin një karrocë me rrota rreth moshës 45 vjeç.
Megjithëse FA nuk mund të shërohet, trajtimet adekuate simptomatike (si trajtimi i problemeve të zemrës, diabetit dhe ndërhyrjet ortopedike) ndihmojnë në zvogëlimin e ashpërsisë së komplikimeve dhe rreziqeve të tjera që lidhen me sëmundjen dhe më pas zgjasin jetëgjatësinë e pacientëve.
Si ndikon FA në trup?
FA fillon në pjesën e pushtme të trupit, pastaj përhapet në pjesën e sipërme, duke përfshirë krahët dhe duart, dhe pastaj trungun. Dëmtimi gjenetik ndikon gjithashtu sytë, dëgjimin dhe të folurin. Probleme të tjera që lidhen me FA përfshijnë lakim të rëndë anormal të shpinës (scoliosis), sëmundje të zemrës dhe diabet. FA nuk ndikon inteligjencën.
Humbja e koordinimit dhe dobësia e muskujve shkakton probleme me ekuilibrin, duke e bërë një person të paqëndrueshëm dhe të prirur të bjerë. Muskujt e dobët të fytit mund të shkaktojnë vështirësi në të folur dhe në gëlltitje. Dëmtimi i nervit mund të shkaktojë lëvizje të pavullnetshme të syve dhe humbje të dëgjimit. Njerëzit me FA shpesh përjetojnë lodhje, qëndrueshmëri të ulët dhe ngurtësi muskulore. Shumë njerëz me FA përfundimisht zhvillojnë sëmundje të zemrës, të cilat mund të shkaktojnë frymëmarrje, dhimbje gjoksi dhe ritme jonormale të zemrës. Deri në 20% e pacientëve gjithashtu zhvillojnë diabetin.
Si trajtohet FA nga mjekësia konvencionale?
Aktualisht nuk ekziston asnjë kurë kimike për FA, kështu që trajtimi konsiston në menaxhimin e simptomave. Ndihma për këmbët, karriget me rrota dhe terapitë fizike ose profesionale mund të ndihmojnë me lëvizjen dhe forcën e muskujve. Medikamentet mund të kontrollojnë disa simptoma të sëmundjeve të zemrës. Ilaçet e insulinës ose të uljes së glukozës mund të kontrollojnë diabetin dhe kirurgjia mund të korrigjojë lakminë e shpinës ose deformimet e këmbëve. Terapia e të folurit dhe këshillimi emocional mund të jenë gjithashtu të dobishme.
Kërkimi për trajtime më të mira është në vazhdim, me shumë studime kërkimore në zhvillim, disa që përpiqen të zbulojnë detajet rreth asaj që shkakton FA, dhe të tjerët duke kërkuar trajtime të reja.
Trajtimi Natyral Potencial për Friedreich Ataxia
Friedreich Ataxia rezulton nga modifikimet në sekuencat e ADN-së që parandalojnë qelizat nga prodhimi i një proteine të nevojshme të quajtur frataxin. Mungesa e frataxin mund të rezultojë në një shumëllojshmëri të problemeve që përfshijnë humbjen e kontrollit të muskujve, lodhjen, dëmtimin e shikimit ose të dëgjimit, fjalimit të ngathët dhe kushteve të rënda të zemrës.
Duke përdorur kurën bimore të FA, hulumtimet kanë identifikuar një mënyrë për të lejuar rifillimin e prodhimit normal të frataxinës.
Kura bimore për FA ka efekt në parandalimin e sekuencës së mutantëve nga përkulja dhe bllokimi i gjenit frataxin. Ky veprim aktivizon gjenin frataxin, i cili pastaj i vendos frataxinat RNA dhe proteinat në nivele normale. Përveç kësaj, qasja jonë është selektive për targetimin e gjenit Frataxin FXN dhe nuk ndikon në gjenet e tjera.
Kura vepron në mënyrë natyrale dhe me shumë potencial për parandalimin e agresivitetit të sëmundjes dhe problemeve që ajo shkakon me zemrën, diabetin, shikimin dhe dëgjimin, lëvizjen dhe përkuljen e trungut, duke përmirësuar në maksimumin e mundshëm cilësinë e jetës së pavientëve me një prognozë shumë premtuese.